
Genomics: the end of the beginning
The cells of every living organism contain a set of fundamental instructions – the genome. The genome consists of long DNA molecules that form chromosomes, which provide the information the cell needs in order to operate. Understanding our genome means understanding our genetic identity which sets us apart from other individuals and other species.
Enter the genomic era. In 2001, the draft sequence of the human genome was completed after more than a decade of intentional effort and a cost of 2.7 billion dollars. This landmark achievement of sequencing and assembling nearly 3 billion basepairs was not the beginning of the end, but the end of the beginning. Since then, incredible advances in DNA sequencing technologies have started to revolutionize biology. Today, generating a draft human genome sequence can cost around $1000 and can be completed in days. In parallel, the remarkable adoption of the CRISPR/Cas bacterial immune system as an efficient DNA-editing tool has made it easy to create new genomic sequences. Now, the stage is set to take on the monumental challenge of deciphering the secrets of the genome.
Genome structure and function
The cells of our body share the same genome and thus the same set of genes. Yet a muscle cell, a blood cell and a brain cell are all very different from each other in shape and function. How is this possible? Rather than constantly activating all their genes, our cells operate somewhat like a computer (or a mathematical function): based on its current condition and on input from its surroundings, a cell calculates how to act, resulting in activation of a specific set of appropriate genes. This complex regulatory system which performs the calculation gives the cells their unique functional identity, and understanding how it works is one of the central goals of modern biology.
Interestingly, gene regulation is closely linked to the spatial organization of the genome. For example, special regulatory sequences can physically interact and control the activation of genes that are far from them in the genomic sequence. It is thus not surprising that the 3D organization of the genome is not random, but hierarchical and ordered. In fact, the spatial organization of the genome is closely linked to most nuclear processes, both natural and in disease, in a wide range of biological species.
What we study
The two main fundamental questions we study are:
- How is the 3D organization of the genome encoded?
- How does the 3D organization of the genome mediate its function?
How we study it
We use high-throughput experiments, mainly based on next-generation DNA sequencing technology. One of the central techniques we use is Hi-C, an experiment that allows to measure pairs of loci in the genome that contact each other in 3D, at a single point in time, across the entire genome. After performing these experiments in our lab, measuring hundreds of millions of interacting pairs in a single experiment, we construct interaction maps – matrices that show how frequently every pair of loci in the genome interacts.
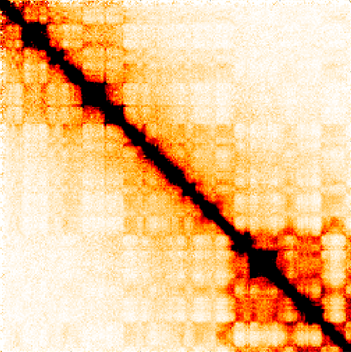
Hi-C interaction map
Some of the major challenges in this field include using these interaction maps to computationally identify structural patterns, analyzing these patterns, and associating them with other functional genomic data. We use a variety of computational approaches, with a focus on machine learning and probabilistic models. However, we try to go beyond simple descriptive and correlative analysis, to produce quantitative predictive models that incorporate explicit assumptions about the underlying biological mechanisms. We can then test our models in the lab by performing genetic perturbations and genomic assays. By studying a diverse set of biological systems, we aim to uncover general principles underlying genome structure and function.